

{kind=link}
Un nuevo dispositivo fue avalado por la FDA en Estados Unidos. Aunque no es de uso masivo ni preventivo, es un paso clave para identificar signos tempranos de la enfermedad que representa entre el 60% y el 70% de los casos de demencia.

La Administración de Alimentos y Medicamentos (FDA) de Estados Unidos, aprobó por primera vez un test sanguíneo in vitro diseñado específicamente para evaluar la enfermedad en pacientes con deterioro cognitivo. Se trata de una prueba llamada Lumipulse G pTau 217/β-Amyloid 1-42 Plasma Ratio, que fue incorporada en todos los laboratorios clínicos del país. La validación oficial marca un cambio de paradigma: a partir de ahora, una simple muestra de sangre permitirá a los médicos identificar signos tempranos y la progresión del Alzheimer sin necesidad de recurrir, en primera instancia, a procedimientos más invasivos como las punciones de líquido cefalorraquídeo o los escáneres cerebrales por tomografía.
“La aprobación de hoy es un paso importante para el diagnóstico del Alzheimer, lo que facilita el acceso más temprano a esta herramienta para los pacientes en Estados Unidos”, señaló la doctora Michelle Tarver, directora del Centro de Dispositivos y Salud Radiológica de la FDA.
El dispositivo, desarrollado por Fujirebio, no apunta al uso masivo ni preventivo: está indicado para adultos mayores de 50 años que presenten síntomas de deterioro cognitivo y que ya estén siendo evaluados por un equipo médico especializado. En estos casos, la prueba actúa como un primer paso para confirmar o descartar la presencia de placas β-amiloides en el cerebro, una de las señales biológicas más reconocidas del Alzheimer.

La nueva herramienta se inserta en un escenario donde el diagnóstico suele llegar tarde. La enfermedad de Alzheimer se desarrolla a lo largo de años, y en algunos casos incluso décadas, antes de que los síntomas se manifiesten de manera clara. La falta de accesos diagnósticos accesibles y no invasivos dejó durante mucho tiempo a millones de pacientes sin una evaluación precisa en fases iniciales, cuando las intervenciones disponibles pueden tener mayor impacto.
Los investigadores ven en este nuevo test una forma de mejorar el acceso, reducir la incertidumbre clínica y habilitar rutas de tratamiento más ajustadas al perfil del paciente. Para la FDA, la autorización marca un hito regulatorio: hasta ahora, existían pruebas similares desarrolladas y comercializadas sin su aval, bajo una figura legal conocida como “tests desarrollados en laboratorios”.
Esta categoría, menos supervisada, permitió que laboratorios privados ofrecieran servicios de diagnóstico sin validación suficiente, lo que generó inquietud entre médicos y asociaciones de pacientes. La aprobación de Lumipulse propone un nuevo estándar.
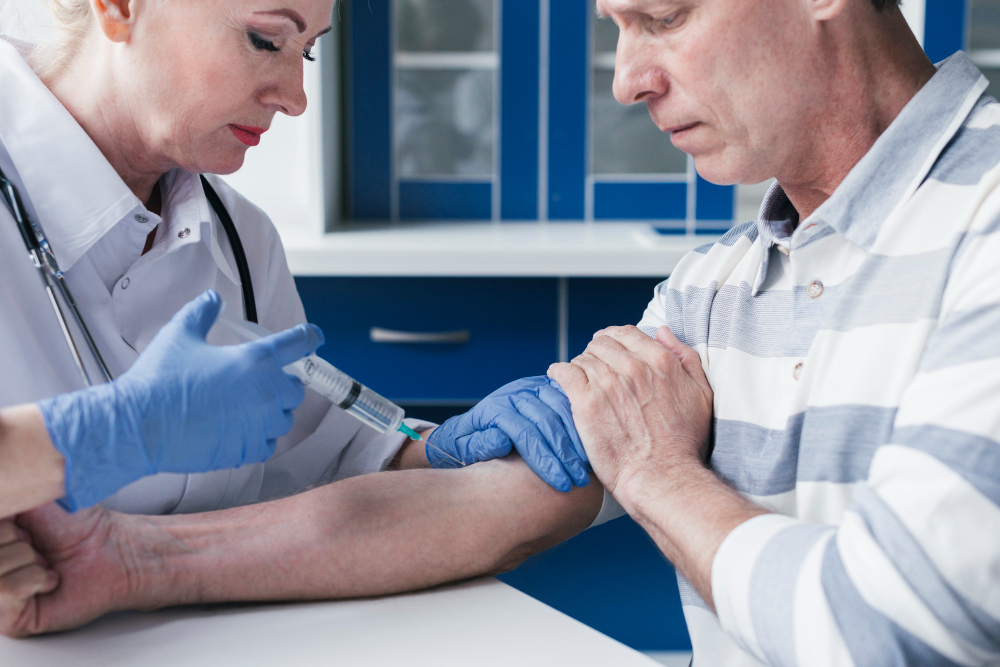
Según el estudio que respaldó la solicitud ante la FDA, el test fue probado en una población de 499 pacientes con signos de deterioro cognitivo. Los resultados mostraron un valor predictivo positivo del 92% y un valor predictivo negativo del 97%, es decir, un nivel de concordancia muy alto con los métodos tradicionales de diagnóstico. Solo el 20% de los casos quedó en zona gris, lo que implicó la necesidad de estudios adicionales.
La enfermedad de Alzheimer actúa en al cerebro dañando sus componentes más básicos: las neuronas. Hace que no cumplan su función y finalmente mueran. El proceso por el cual esta enfermedad destruye las neuronas se llama neurodegeneración. Sus síntomas son cognitivos y conductuales, tales como problemas de memoria, de orientación y confusión y es la causa más común de demencia en adultos mayores.
Según la Organización Mundial de la Salud (OMS), la enfermedad de Alzheimer es la forma más común de demencia y puede representar entre un 60% y un 70% de los casos.
Si bien el Alzheimer no tiene cura, existen fármacos que permiten mitigar los síntomas, retrasar la progresión y mejorar la calidad de vida. Pero su efectividad es limitada en estadios avanzados.
Mejores decisiones clínicas
El avance validado por la FDA es más que un logro técnico: representa un cambio en la forma de entender el abordaje clínico de las enfermedades neurodegenerativas. El nuevo test se realiza con una muestra de sangre y puede procesarse en equipos automatizados como el Lumipulse G1200, que ya está operativo en numerosos laboratorios de Estados Unidos.
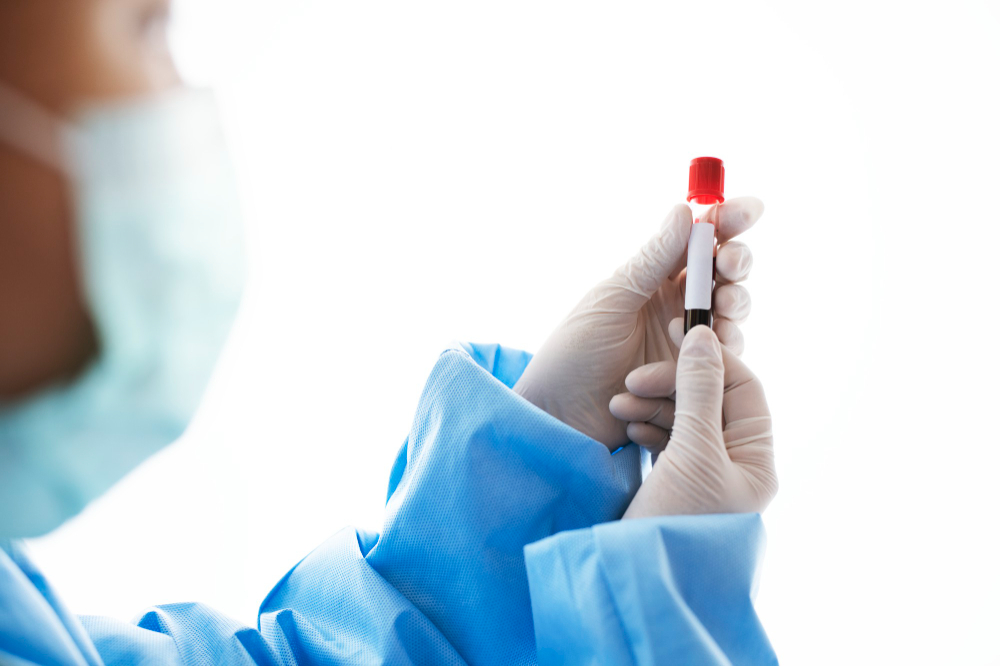
Este sistema automatizado puede procesar hasta 120 pruebas por hora, lo que transforma su aplicación en una solución viable dentro de la rutina de los servicios clínicos especializados. Este volumen de procesamiento acorta los tiempos de espera para los resultados, lo que en muchos casos puede definir el inicio o no de una estrategia terapéutica.
La decisión de la FDA no implica que el nuevo test sustituya por completo los métodos anteriores. La prueba no reemplaza las imágenes cerebrales ni los análisis de líquido cefalorraquídeo. Sin embargo, se posiciona como un filtro previo de gran precisión y bajo costo que permite orientar los pasos siguientes en el diagnóstico. En ese sentido, podría evitar exámenes innecesarios o servir para seleccionar a qué pacientes conviene derivar a estudios más complejos. Para los médicos, representa una herramienta de apoyo diagnóstico. Para los pacientes, una opción menos invasiva y más accesible.
En paralelo al avance clínico, la aprobación de la prueba también tuvo un impacto regulatorio. Durante los últimos años, el gobierno de Estados Unidos buscó aumentar el control sobre los test desarrollados en laboratorios independientes. El argumento oficial fue claro: sin validación suficiente, los pacientes pueden recibir diagnósticos incorrectos y quedar fuera de tratamientos que podrían haber mejorado su evolución. No obstante, en marzo pasado un juez federal de Texas anuló una norma que intentaba restringir el uso de este tipo de test. La demanda fue impulsada por la Asociación Estadounidense de Laboratorios Clínicos, que consideró que la medida afectaría la operatividad del sistema.
“La normativa de la FDA fue diseñada hace medio siglo, cuando los test creados y utilizados por un solo laboratorio eran simples, de bajo volumen, y orientados a necesidades locales”, explicaron voceros sanitarios.
Hoy, en un contexto donde las enfermedades neurodegenerativas ganan espacio en las prioridades de salud pública, el desarrollo de pruebas con validez científica cobra otra dimensión. Por eso, la aprobación de la prueba se interpretó también como una señal: el diagnóstico del Alzheimer entra en una nueva etapa de precisión, rapidez y validación oficial.
La comunidad científica, los pacientes y las organizaciones que acompañan a quienes viven con Alzheimer recibieron la noticia con optimismo. Las demencias afectan hoy a más de 55 millones de personas en el mundo. Cada año se diagnostican 10 millones de nuevos casos. La mayoría corresponden a Alzheimer, una patología que representa entre el 60% y el 70% del total. Detectarla a tiempo no garantiza una cura, pero permite decidir mejor.